The FunFOLD2 Protein-Ligand Binding Site Prediction Server
NOTE: This server is no longer maintained, please use the latest version of IntFOLD to obtain FunFOLD predictions.
Docker package for FunFOLD5
About the latest server
The FunFOLD2 server provides:
- A list of residues in your target sequence that are most likely to bind a ligand
- A list of putative binding ligands
- 3D models of the likely protein-ligand interactions
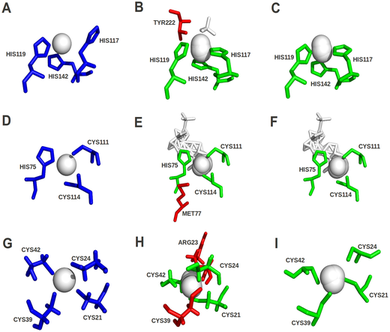
Quality assessment has now been integrated into version 2.0 of the FunFOLD server to improve prediction selection. Figure taken from Roche et al. (2012). Quality assessment scores for ligand binding sites can be used to improve predictions. The predictions centre column were made before quality checks and the predictions in the right column were made after checks were implemented. The green sticks represent residues in the model that were correctly predicted as binding to the ligands. The red sticks represent residues that were not predicted or incorrectly predicted as ligand binding residues. The blue sticks represent the observed ligand binding site residues in the experimental structure.
FunFOLD server version 2.0
FunFOLD server version 1.0
Download standalone versions of FunFOLD and FunFOLDQA
Limitations of the method
Predicting ligand binding sites is a difficult task and there are several limitations to current prediction methods. The following is a list of the most common limitations specific to the current implementation of the FunFOLD2 server: 1. If the server is unable to build a starting model for the target sequence then it cannot predict any ligand binding sites. Although, fortunately for the majority of protein targets a reasonable 3D model can be obtained; 2. If no structural similarity can be found between the target model and structurally elucidated proteins with bound biologically relevant ligands, then a prediction is not made; 3. Only one ligand binding site is predicted per target sequence - the site with the largest identified ligand cluster. However, the server does also provide the data showing all putative ligand clusters and these clusters can be made visible to users on the results page using the Jmol plugin; 4. The FunFOLD2 server currently outputs predictions based on the best predicted IntFOLD2-TS model but the top predicted model may not always be the best model.
News
- Jun 2013: FunFOLD2 server paper published in NAR.
- Dec 2012: FunFOLD2 server now online and open to all users.
- Dec 2012: Success at CASP10: FunFOLD2 among top ranked methods; short talk about FunFOLD2 given at the function prediction session.
- Feb 2011: Original version of FunFOLD method described and accepted for publication.
- Dec 2010: Success at CASP9: FunFOLD among top ranked methods.
References
The FunFOLD2 server reference:- Roche, D. B., Buenavista, M. T., McGuffin, L. J. (2013) The FunFOLD2 server for the prediction of protein-ligand interactions. Nucleic Acids Res., 41, W303-7. PubMed The FunFOLD2 server makes use of the FunFOLDQA Quality Assessment protocol for improved prediction selection, which is described in the following paper:
- Roche, D. B., Buenavista, M. T. & McGuffin, L. J. (2012) FunFOLDQA: A Quality Assessment Tool for Protein-Ligand Binding Site Residue Predictions. PLoS ONE, 7, e38219. PubMed Starting 3D models for FunFOLD2 are built using the novel IntFOLD2-TS protocol, which is described in the following paper:
- Buenavista, M. T., Roche, D. B. & McGuffin, L. J. (2012) Improvement of 3D protein models using multiple templates guided by single-template model quality assessment. Bioinformatics, 28, 1851-1857. PubMed The original paper describing FunFOLD version 1.0:
- Roche, D. B., Tetchner, S. J. & McGuffin, L. J. (2011) FunFOLD: an improved automated method for the prediction of ligand binding residues using 3D models of proteins. BMC Bioinformatics, 12, 160. PubMed