The DomFOLD Protein Domain Boundary Prediction Server
NOTE: This server is no longer maintained, please use the latest version of IntFOLD to obtain DomFOLD predictions.
About the server
The DomFOLD Protein Domain Prediction Server allows you to predict the number of domains and their possible boundaries within a protein sequence. Two methods are available: DomFOLD and DomFOLDpdp. In the DomFOLD method, the output from DomSSEA, DISOPRED and HHsearch is parsed to form a consensus domain prediction (Figure 1). In the DomFOLDpdp method, the top fold recognition model from nFOLD3 is analysed using the program PDP (Figure 2).
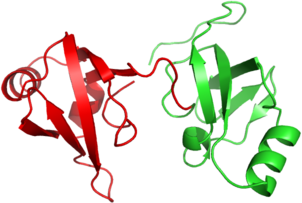
Figure 1 DomFOLD prediction for the hard 2 domain CASP8 target T0462. The native structure has been coloured in accordance with the domain boundary predicted by DomFOLD during the CASP8 experiment. The image was rendered using Pymol.
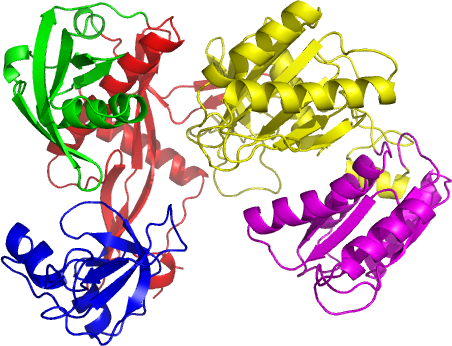
Figure 2 DomFOLDpdp prediction for the 5 domain CASP8 target T0487. The native structure has been coloured in accordance with the domain boundaries predicted by DomFOLDpdp. The image was rendered using Pymol. N.B. DomFOLDpdp is a new method and did not participate in CASP8, however the nFOLD3 model used by DomFOLDpdp was a blind prediction.
DomFOLD Submission Form
News
- The latest DomFOLD method is now integrated with the IntFOLD server.
- September 2008: Version 2.0 of the server is now online, which includes the new DomFOLDpdp method.
- March 2008: The new DomFOLD server will be available in time for CASP8.
References
The DomFOLD method incorporates the following methods: DomSSEA, DISOPRED and HHsearch
- Marsden, R., McGuffin, L. J. & Jones, D. T. (2002) Rapid protein domain assignment from amino acid sequence using predicted secondary structure. Protein Science, 11, 2814-2824. PubMed
- Ward, J. J., Sodhi, J. S. McGuffin, L. J., Buxton, B. F. & Jones, D. T. (2004) Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol., 337, 635-645. PubMed
- Söding J. (2005) Protein homology detection by HMM-HMM comparison. Bioinformatics. 21, 951-96. PubMed
The DomFOLDpdp method uses the PDP program:
- Alexandrov, N. & Shindyalov, I. (2003) PDP: protein domain parser. Bioinformatics. 19, 429-30. PubMed