The DISOclust Protein Disorder Prediction Method
NOTE: This server is no longer maintained, please use the latest version of IntFOLD to obtain DISOclust predictions.
About the server
The DISOclust method provides predictions of protein disorder based on the analysis of 3D structural models using ModFOLDclust. The DISOclust results are combined with those from an in-house version of DISOPRED in order to improve predictions.
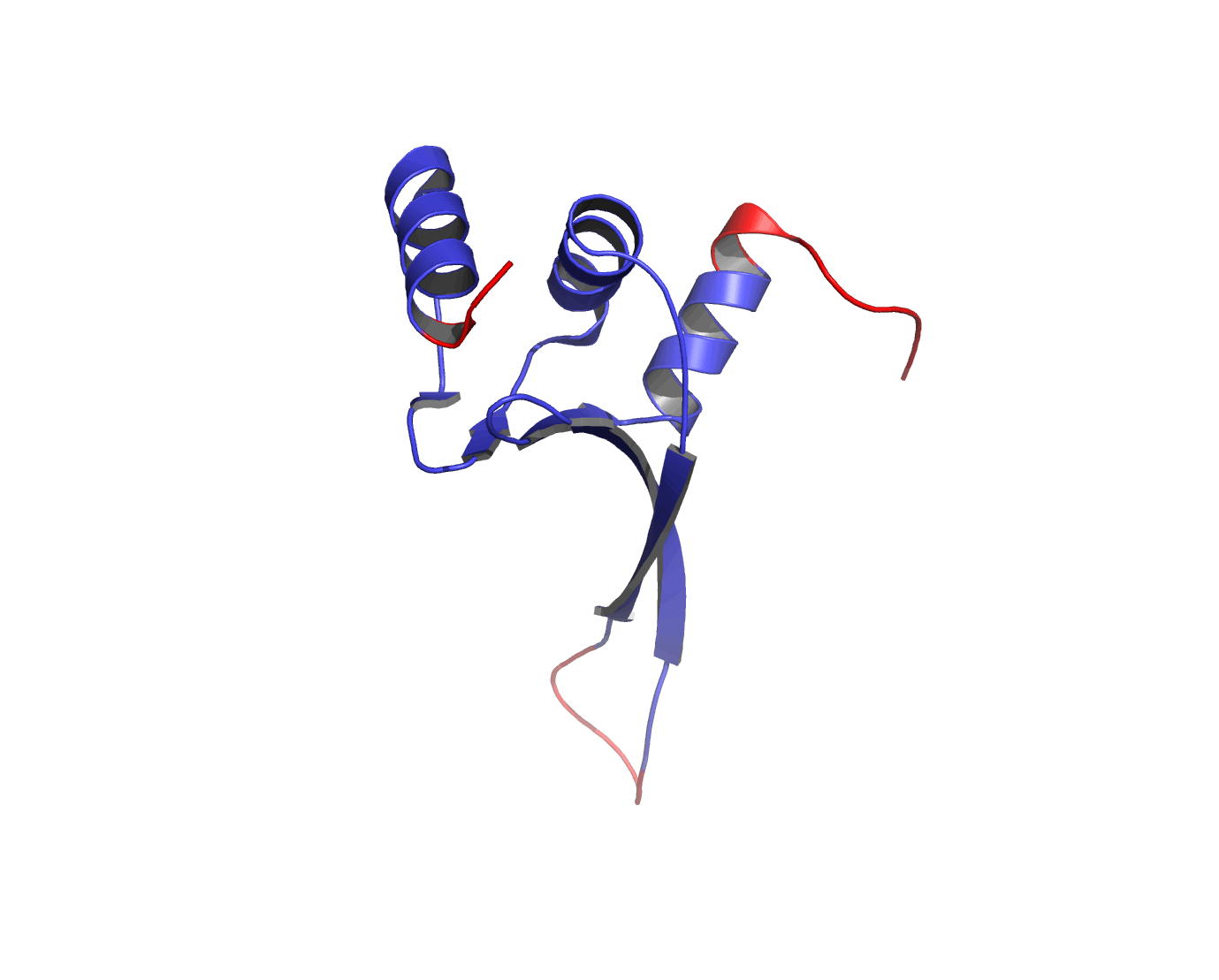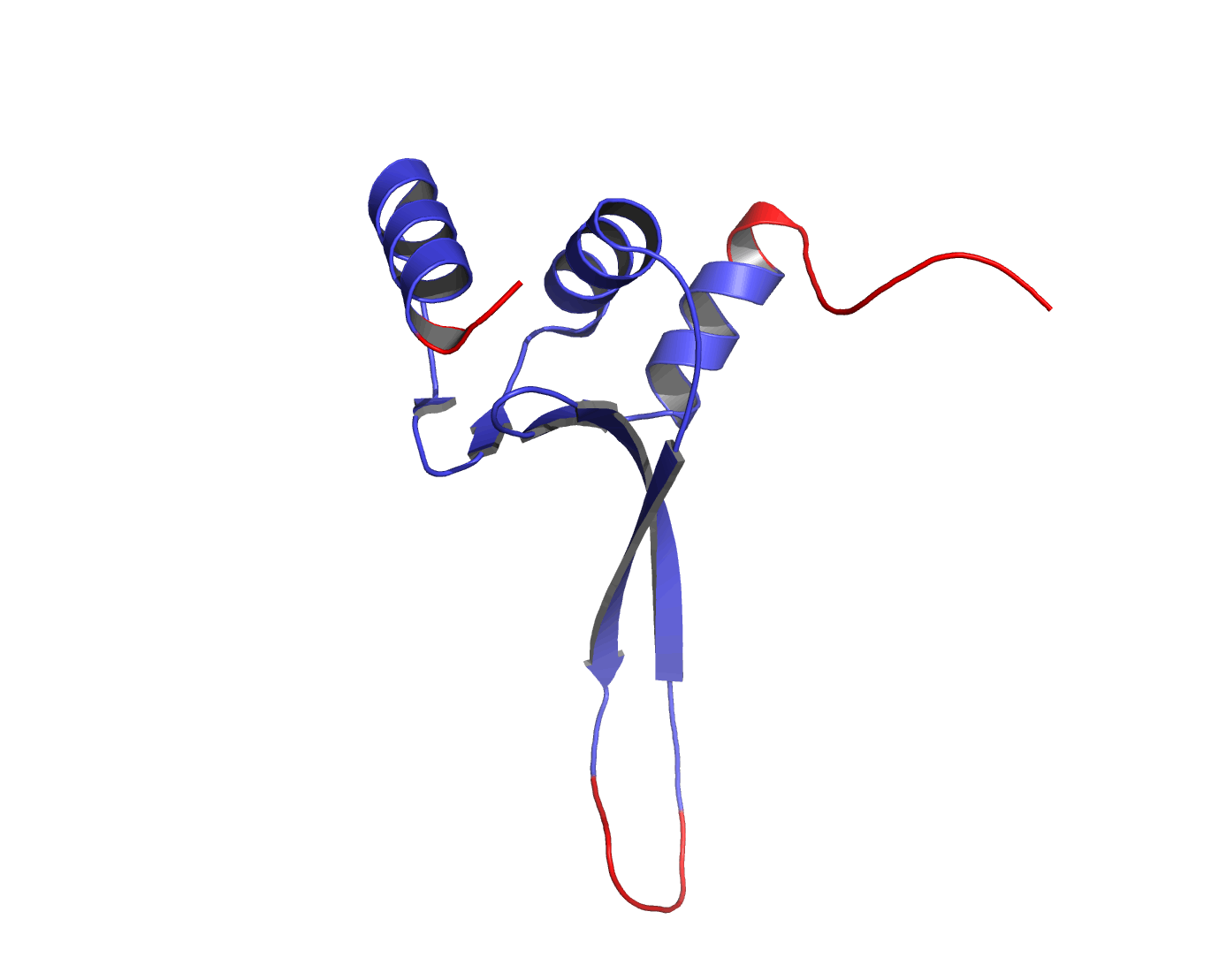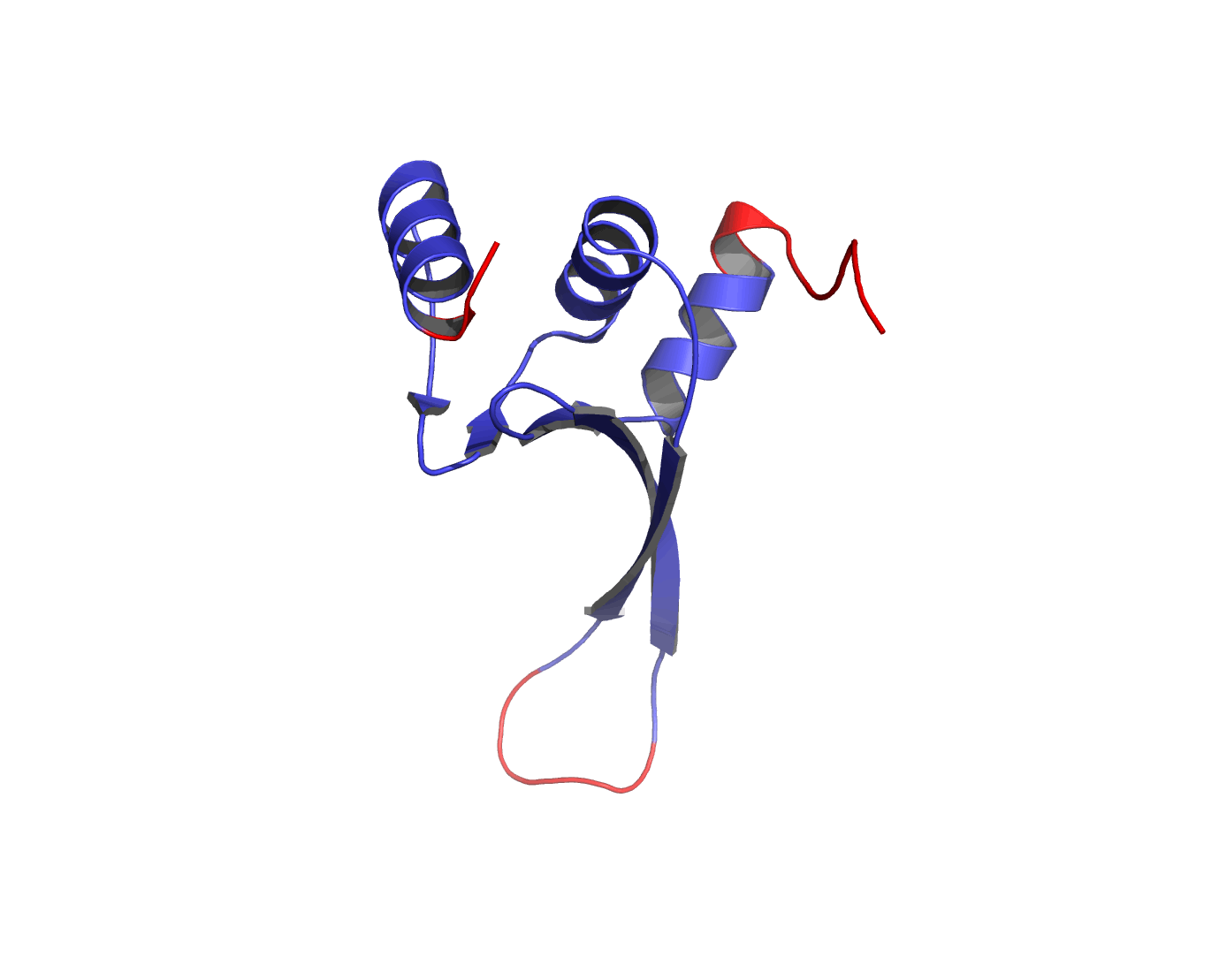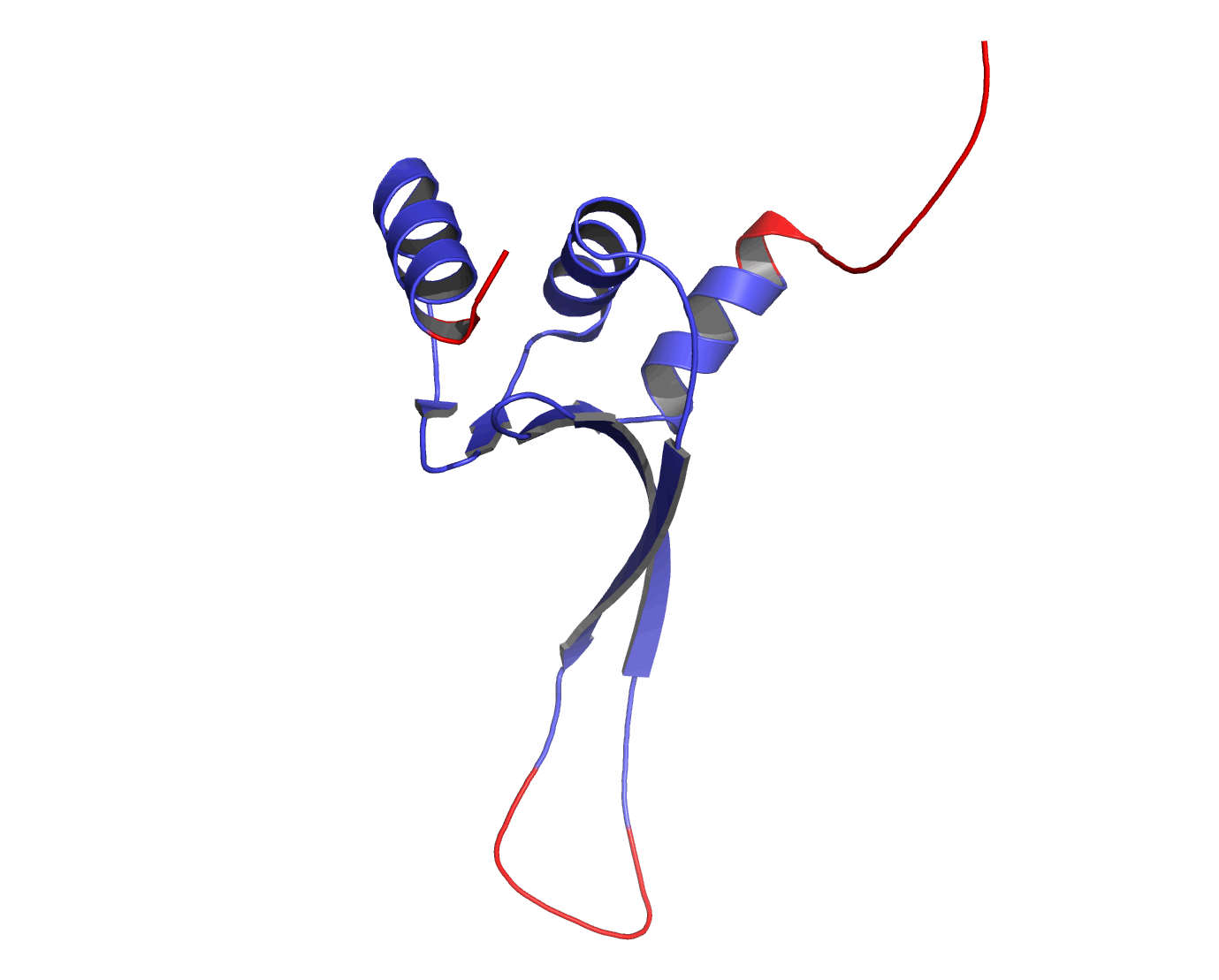
Figure 1The DISOclust predicted disorder is mapped onto an ensemble of NMR models for CASP target T0460 (PDB code 2k4n). Click on the image to see an animation showing individual NMR models. The residues predicted to be disordered are coloured red; blue residues are predicted to be ordered. The image was rendered using Pymol (http://www.pymol.org).
The DISOclust Server v 1.1
Download DISOclust
Obtain DISOclust predictions via the ModFOLD server (use your own models)
News
- The latest DISOclust version is now integrated with the IntFOLD server.
- Jan 2009: Version 1.1 of the DISOclust server is now online. Submit your target sequence and receive intuitive graphical results.
- Dec 2008: DISOclust is one of the top performing methods at CASP8
- March 2008: Prototype DISOclust server ready for testing at CASP8.
References
Please cite the following paper when refering to the original description of the DISOclust method:- McGuffin, L. J. (2008) Intrinsic disorder prediction from the analysis of multiple protein fold recognition models. Bioinformatics, 24, 1798-804. PubMed
- McGuffin, L. J. (2008) The ModFOLD Server for the Quality Assessment of Protein Structural Models. Bioinformatics, 24, 586-587. PubMed
- Ward, J. J., Sodhi, J. S., McGuffin, L. J., Buxton, B. F. & Jones, D. T. (2004) Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol., 337, 635-645. PubMed